· The Secretary of Defense only has the authority to mandate the administration of a fully Approved, Licensed, and Appropriately Labeled “Biological Product.”
· An approved biological product is licensed by the Department of Health and Human Services (HHS), and are required to have the HHS License No., Product Trade Name, Manufactures Name, & Address plainly marked on the label.
· Currently, the only available Covid-19 vaccines all exist under Emergency Use Authorization (EUA) status. EUA products are mutually exclusive in relationship to licensed and available biological products, they cannot exist together.
· Only the President of the United States can waive and mandate the option to refuse an EUA product to DoD personnel, and must do so in writing, and for a very specific and constrained reason.
· There are no interchangeable drugs that are approved in lieu of the approved HHS No. 2229 (Comirnaty) and HHS No. 2256 (Spikevax). Interchangeability is statutory, and clearly defined in DoDI 6200.02, and only involves two or more licensed biological products (To-Date there are only two interchangeable drugs in the US;Cyltezo & Humira).
· BLA-Compliant is a made-up term that holds no legal basis in terms of DoD ability to mandate, nor does it have any medical basis in terms of regulation of Biological Products by the federal government.
Pertinent & Incontrovertible Facts:
1. The 23 August 21 FDA approved (BLA #125742), HHS licensed (No. 2229) BioNTech COMIRNATY vaccine (NDC 0069-1000-XX), nor the 16 December 21 addition (NDC 0069-2025-XX) to prevent Covid-19 in individuals 16 years of age and older is UNAVAILABLE in the US.
2. NDC 0069-1000-XX & NDC 0069-2025-XX are not in production, nor are they planned to go into production until exhaustion of the EUA vaccines.
3. The 31 January 22 FDA approved (BLA #12572), HHS licensed (No. 2256) ModernaTX SPIKEVAX vaccine (No NDC available) to prevent Covid-19 in individual 18 years of age and older, is UNAVIALABLE in the US.
4. Due to the licensed vaccines (No. 2229 & No. 2256) not being available to the military, or the general public, the FDA has been forced to reauthorized similar versions under Emergency Use Authorization.
5. The unapproved EUA vaccines are “legally distinct”, and cannot considered biosimilar, or interchangeable according to statute. This is confirmed in the FDA Purplebook. Unapproved EUA vaccines do not contain the statutorily required approval HHS License Number, Trade Name, Manufacturing Address, or licensed NCDs
6. The only available vials manufactured to prevent COVID-19 available in the US are manufactured and labeled for “use under Emergency Use Authorization” They also lack any HHS License Numbers and have the following unlicensed NDC#s: 59676-580-05 (Janssen), 80777-273-10 (Moderna), 59267-1000-1, 59267-1055-1, 59267-0078-1, & 52967-1025-1 (Pfizer-BioNTech). EUA vaccines, and Approved vaccines are mutually exclusive if the Approved is available
7. In order to be considered the BLA approved vaccine, the label is required to contain the trade name, US license No. and manufacturing locations. (Public Health Act in Section 351(a)(1)(B))
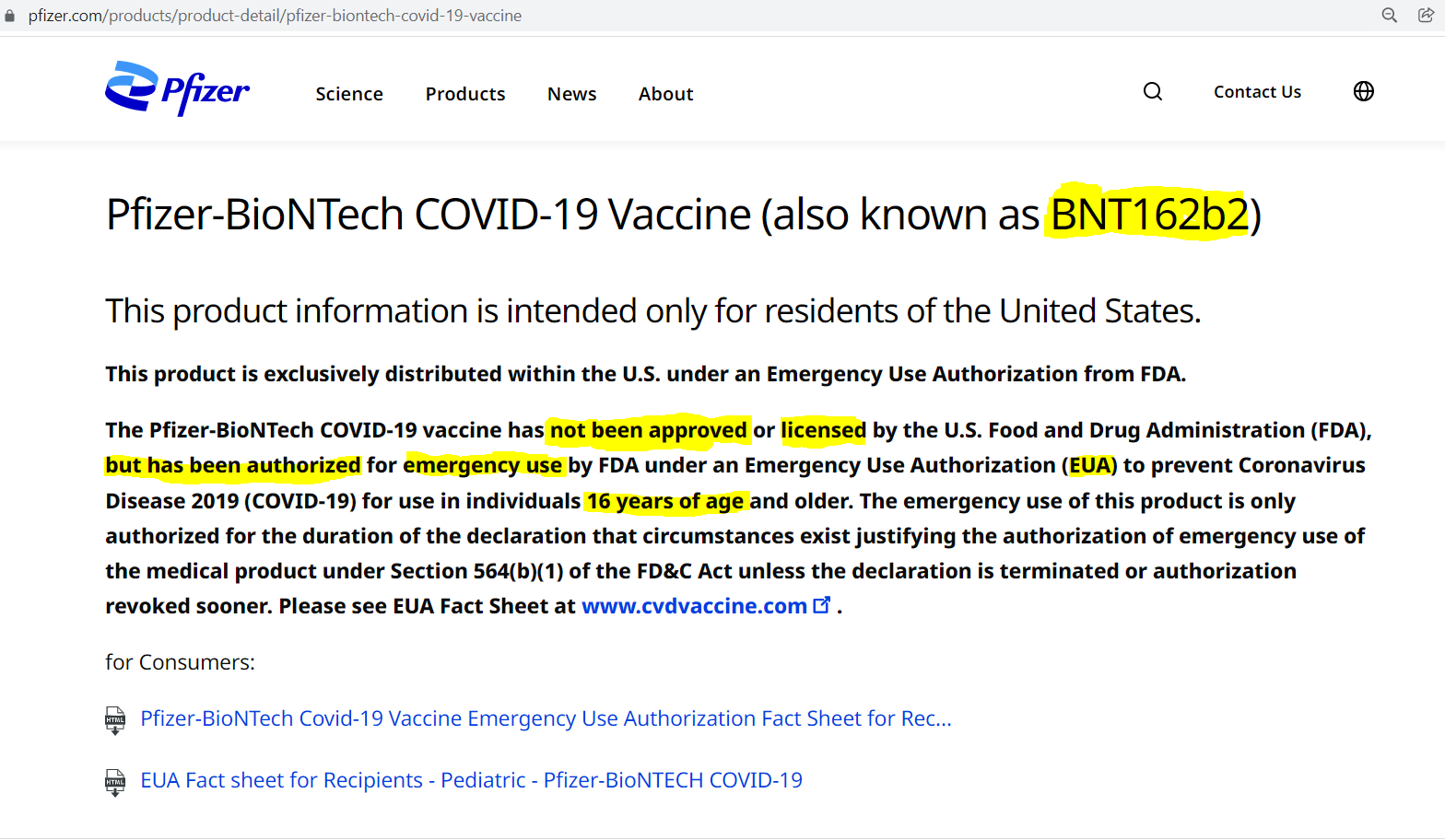
8. Pfizer states it’s Pfizer-BioNTech Covid-19 vaccine (BNT162B2) is not FDA approved or licensed, but merely authorized under EUA for individuals 16 years of age and up, on its own website
9. The FDA, CDC, and NIH all state that the licensed vaccines (No. 2229 ‘COMIRNATY’ & No. 2256 ‘SPIKEVAX’) are not available
10. Mandates on military members currently require that vaccines are “accordance with FDA-approved labeling and guidance”
11. Without access to the approved vaccine, it is impossible for the Secretary of Defense or any subsequent officer order military personnel by mandate, because in order to become compliant it requires voluntary inoculation with EUA vaccines.
12. Current US & Military regulations have provisions for this exact scenario:
a. Waiver by the President to mandate the EUA vaccines
b. In absence of Presidential waiver of EUA, a medical exemption is available due to lack of approved vaccine supply (MS) – 90 day waiver.
What did the President’s Lawyers recommend the President do?
“As for DOD’s concern about service members who would lack a meaningful option to refuse EUA products because of the prospect of sanction, including possibly prosecution, we note that any difference between our view and the assumption reflected in the conference report should have limited practical significance. Given that FDA has imposed the “option to accept or refuse” condition for the COVID-19 vaccines by requiring distribution of its Fact Sheet containing the “[i]t is your choice to receive or not receive” language, DOD is required to provide service members with the specified notification unless the President waives the condition pursuant to 10 U.S.C. § 1107a. And because DOD has informed us that it understandably does not want to convey inaccurate or confusing information to service members—that is, telling them that they have the “option” to refuse the COVID-19 vaccine if they effectively lack such an option because of a military order—DOD should seek a presidential waiver before it imposes a vaccination requirement.”
What did the Secretary of Defense say mandate?
On 9 August 2021, the Secretary of Defense stated “I will seek the President’s approval to make the vaccines mandatory no later than mid-September, or immediately upon the U.S. Food and Drug Administration (FDA) licensure, whichever comes first. By way of expectation, public reporting suggests the Pfizer-BioNTech vaccine could achieve full licensure early next month.”
On 23 August 2021, the FDA approved BioNTech’s licensed Comirnaty vaccine with a start and end marketing date of 23 August 2021, and subsequently reissued the EUA for the unlicensed Pfizer-BioNTech BNT162 vaccine, specifically because Comirnaty would not be available any time soon.
On 24 August 2021, The Secretary of Defense stated “Mandatory vaccination against COVID-19 will only use COVID-19 vaccines that receive full licensure from the Food and Drug Administration (FDA), in accordance with FDA-approved labeling and guidance.”
What did the Assistant Secretary of Defense advise?
A 14 September 2021 signed memo from Dr. Terry Adirim, Principal Deputy Assistant Secretary of Defense for Health Affairs, stated that: “Per FDA guidance, these two vaccines are “interchangeable” and DoD health care providers should “use doses distributed under the EUA to administer the vaccination series as if the doses were the licensed vaccine. Consistent with FDA guidance”
o Second, the line quoted by Dr. Adirim “The FDA-approved Pfizer-BioNTech product Comirnaty (COVID-19 Vaccine, mRNA) and the FDA-authorized Pfizer-BioNTech COVID-19 Vaccine under EUA have the same formulation and can be used interchangeably to provide the COVID-19 vaccination series without presenting any safety or effectiveness concerns. Therefore, providers can use doses distributed under EUA to administer the vaccination series as if the doses were the licensed vaccine. For purposes of administration, doses distributed under the EUA are interchangeable with the licensed doses. The Vaccine Information Fact Sheet for Recipients and Caregivers provides additional information about both the approved and authorized vaccine.” disappeared and was replaced with: “The FDA-approved Comirnaty (COVID-19 Vaccine, mRNA), made by Pfizer for BioNTech and the FDA-authorized Pfizer-BioNTech COVID-19 Vaccine under EUA have the same formulation and can be used interchangeably to provide the COVID-19 vaccination series without presenting any safety or effectiveness concerns. (XX quote deleted from website XX) For purposes of administration, doses distributed under the EUA are interchangeable with the licensed doses. The Vaccine Information Fact Sheet for Recipients and Caregivers provides additional information about both the approved and authorized vaccines.”
o Compare the 6 September 21 version with the 19 October 21 version:
o Thirdly, the memo from Dr. Adirim reads as the FDA told the DoD that they “SHOULD” use the EUA vaccines as if they were the licensed vaccines. When in reality the guidance said providers “CAN” use, and it is in regard to administration, effectiveness, and safety. Both versions clarify that “The products are legally distinct with certain differences that do not impact safety or effectiveness.”
o Fourthly, not only is it imprudent and calamitous to establish policy off an ever-changing unofficial Q&A website, it is unlawful.
o According to the FDA: “What is the difference between the Federal Food, Drug, and Cosmetic Act (FD&C Act), FDA regulations, and FDA guidance? The Federal Food, Drug, and Cosmetic Act (FD&C Act) is a federal law enacted by Congress. It and other federal laws establish the legal framework within which FDA operates. The FD&C Act can be found in the United States Code, which contains all general and permanent U.S. laws, beginning at 21 U.S.C. 301. FDA develops regulations based on the laws set forth in the FD&C Act or other laws under which FDA operates. FDA follows the procedures required by the Administrative Procedure Act, another federal law, to issue FDA regulations. This typically involves a process known as "notice and comment rulemaking" that allows for public input on a proposed regulation before FDA issues a final regulation. FDA regulations are also federal laws, but they are not part of the FD&C Act. FDA regulations can be found in Title 21 of the Code of Federal Regulations (CFR). FDA follows the procedures required by its "Good Guidance Practice" regulation to issue FDA guidance. FDA guidance describes the agency’s current thinking on a regulatory issue. Guidance is not legally binding on the public or FDA. The Good Guidance Practice regulation can be found at 21 CFR 10.115.”
§ 21 CFR § 10.115 (d)(1)(2)(3): “Are you or FDA required to follow a guidance document? (1) No. Guidance documents do not establish legally enforceable rights or responsibilities. They do not legally bind the public or FDA. (2) You may choose to use an approach other than the one set forth in a guidance document. However, your alternative approach must comply with the relevant statutes and regulations. FDA is willing to discuss an alternative approach with you to ensure that it complies with the relevant statutes and regulations. (3) Although guidance documents do not legally bind FDA, they represent the agency's current thinking. Therefore, FDA employees may depart from guidance documents only with appropriate justification and supervisory concurrence.
o The statutes (BPCI Act, DODI 6200.02, & 42 USC section 262) are clear: to be considered “interchangeable” both drugs must be BLA approved Biological Products, an application of biosimilar, and interchangeability must be submitted and approved, and a 351(k) designation will be listed in the FDA Purplebook
To-Date, there are only two drugs approved and classified “interchangeable” and they are not COVID-19 vaccines
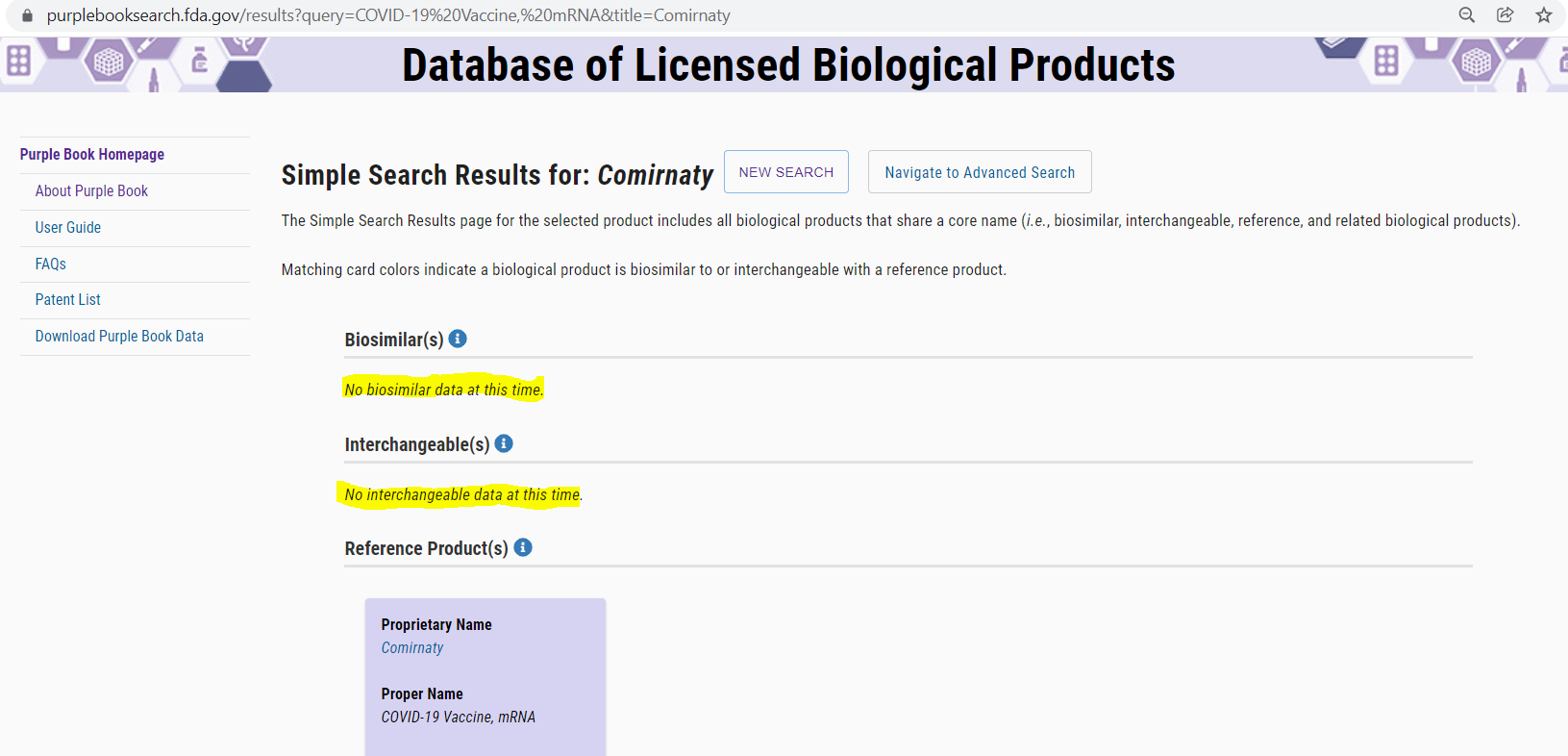
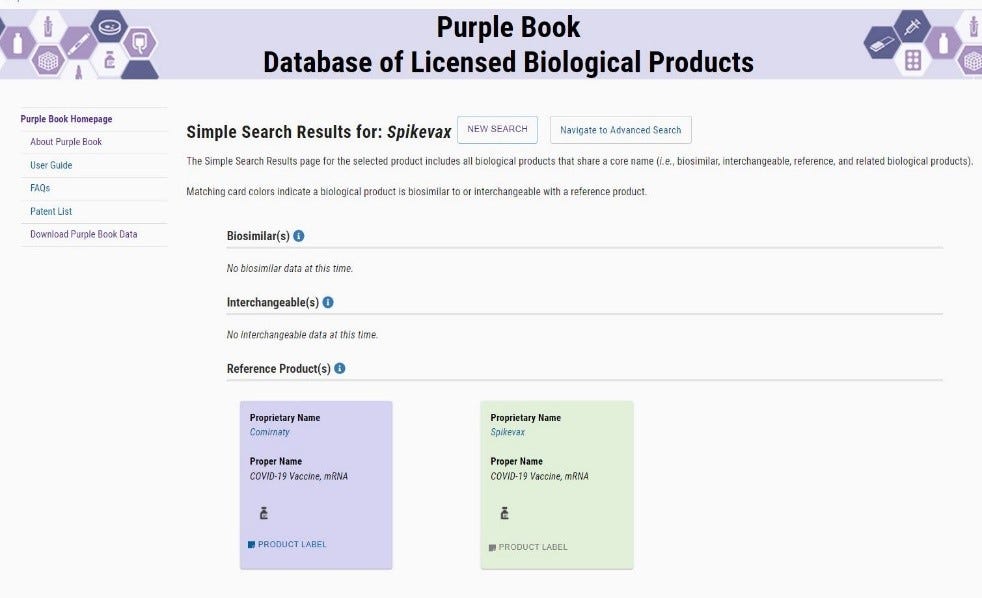
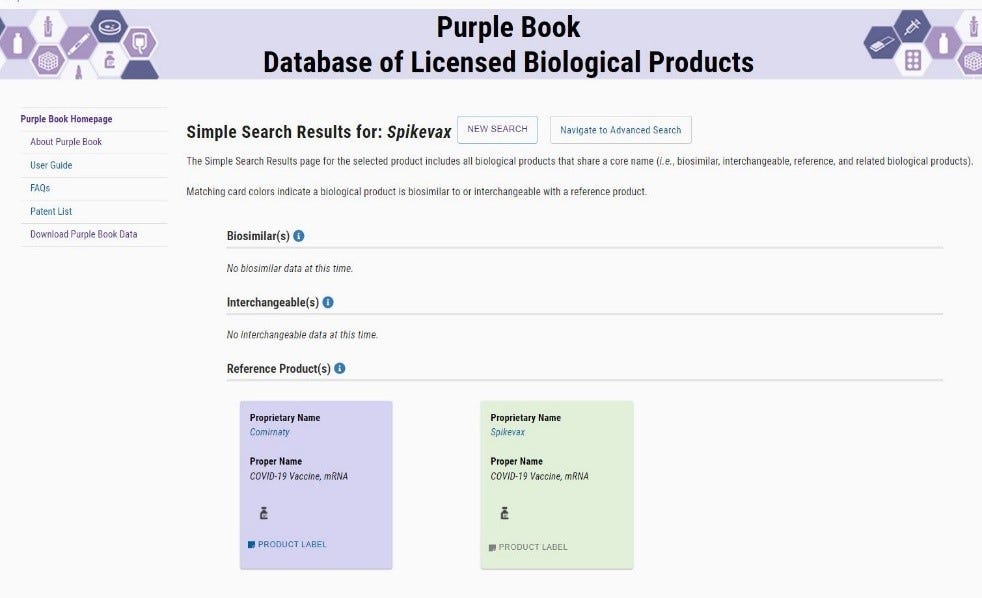
The FDA Purplebook list both Comirnaty & Spikevax as having no biosimilar, or interchangeability with any other vaccines.
o Dr. Adirim’s memo is not only unprofessional from a regulatory standpoint, it is purposefully deceiving, and unenforceable.
What did the FDA approve & what did the FDA authorize?
· On 23 August 2021, The FDA approved BioNTech Manufacturing GmbH’s vaccine Biologics License Application (BLA #125742), HHS US License No. 2229, stating: “You may label your product with the proprietary name, COMIRNATY.” And that: “Content of labeling must be identical to the Package Insert submitted on August 21, 2021”
“Drugs are identified and reported using a unique, three-segment number called the National Drug Code (NDC) which serves as the FDA’s identifier for drugs.” - FDA
o The following NDCs were issued for Comirnaty:
0069-1000-03 (Box of 25 vials)
0069-1000-02 (Box of 195 vials)
0069-1000-01 (Individual vials)
o The Marketing Start and End date for Comirnaty were both 23 August 2021.
o The NIH archived the approved Comirnaty four days later on 27 August 2021.
o The NIH issued a DailyMed Announcement on 13 September 2021 stating: “At present, Pfizer does not plan to produce any product with these new NDCs and labels over the next few months while EUA authorized product is still available and being made available for U.S. distribution. As such, the CDC, AMA, and drug compendia may not publish these new codes until Pfizer has determined when the product will be produced with the BLA labels.
· The approved drug, if produced, would have been required to have a label that looked like this:
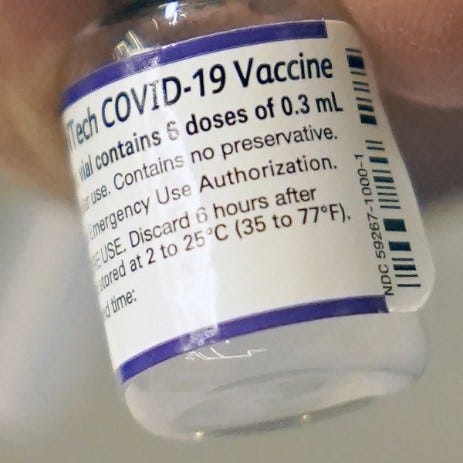
· This allowed administration of the original BNT162 vaccines under EUA, after the approval, but unavailable Comirnaty, with labels that had NDC 59267-1000-1, and continued looked like this:
· Pfizer has also produced vials with the EUA NDC 59267-1000-1 that have purple borders, and cap, however this label is not published on the NIH website. Example:
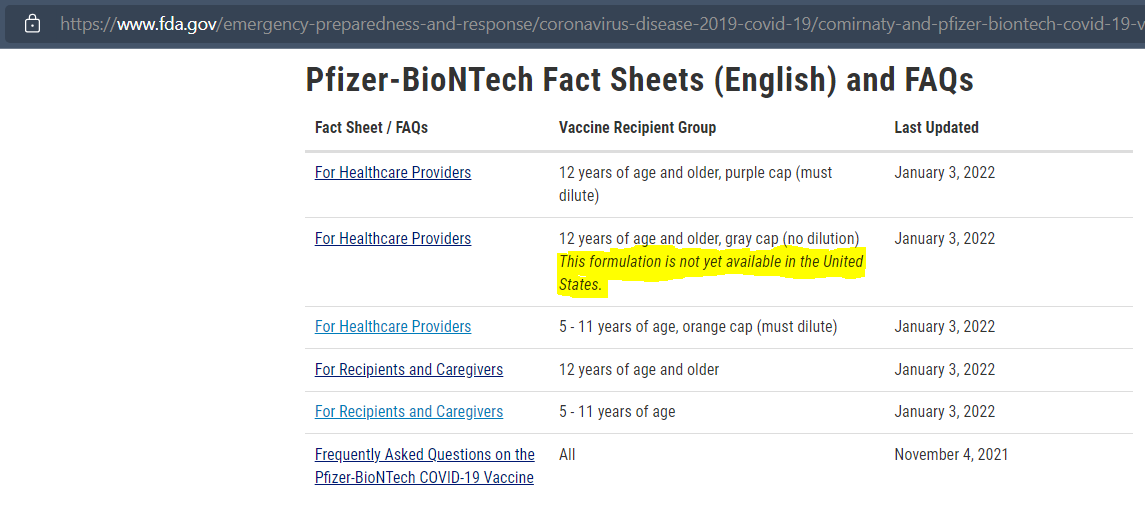
· On 29 October 2021, the FDA authorized a new formulation of the EUA Pfizer-BioNTech with “tromethamine (Tris) buffer instead of phosphate buffered saline (PBS) used in the originally authorized Pfizer-BioNTech COVID-19 Vaccine.” This EUA expanded the authorized age range, and reissued the EUA for individuals 16 and older because the approved drug Comirnaty was still unavailable.
· On 16 December 2021, The FDA expanded the formulation of Comirnaty, which also contained the drug called TRIS (tris(hydroxymethyl)aminomethane) and differentiated by a grey top, stating: “We hereby approve the draft content of labeling including the Package Inserts submitted under amendment 10, dated December 13, 2021, and the draft carton and container labels submitted under amendment 6, dated December 9, 2021.”
· The color scheme mirrored the EUA version (NCD 59267-1025-1), with grey top, also a new NDC was issued (0069-2025-01) and the labeling is required look like this:
· On 3 January 2022, the FDA reissued the EUA for the Pfizer-BioNTech vaccine allowing the EUA versions NDC 0069-1000-1 (blank or purple), & NCD 59267-1025-1 (grey) to still be marketed to populations 16 years old and older. Because “Although COMIRNATY (COVID-19 Vaccine, mRNA) is approved to prevent COVID-19 in individuals 16 years of age and older, there is not sufficient approved vaccine available for distribution to this population in its entirety at the time of reissuance of this EUA.”
· As of 29 January 2022 the FDA website that publishes the documentation related to Comirnaty, and Pfizer-BioNTech has the followingnote:
What does the FDA say about the approved Comirnaty?
“Section 351(a)(1)(B)(ii) of the PHS Act requires that each package of a biological product is plainly marked with, among other things, the applicable license number of the manufacturer of the biological product in order for the biological product to be introduced or delivered for introduction into interstate commerce”
“The PHS Act requires that each “package” of a biological product is plainly marked with, among other things, “the proper name of the biological product contained in the package” and “the name, address, and applicable license number of the manufacturer of the biological product” in order for the biological product to be introduced or delivered for introduction into interstate commerce (see section 351(a)(1)(B) of the PHS Act; 21 CFR 610.61, 610.63, 610.64 and 201.1(m)). The “package” means the “immediate carton, receptacle, or wrapper, including all labeling matter therein and thereon, and the contents of the one or more enclosed containers. If no package, as defined in the preceding sentence, is used, the container shall be deemed to be the package” (21 CFR 600.3(cc)). The “manufacturer” of a biological product regulated under the PHS Act that needs to be identified on each package is the BLA holder (see 21 CFR 600.3(t)(definition of manufacturer); see also 21 CFR 610.63 (labeling requirements for divided manufacturing responsibility)).”
· FDA LOT RELEASE “Please submit final container samples of the product in final containers together with protocols showing results of all applicable tests. You may not distribute any lots of product until you receive a notification of release from the Director, Center for Biologics Evaluation and Research (CBER).”
· “You should identify and investigate all manufacturing deviations promptly, including those associated with processing, testing, packaging, labeling”
· Labeling “The proprietary name, COMIRNATY, was reviewed by CBER’s Advertising and Promotional Labeling Branch (APLB) on July 2, 2021, and found to be acceptable.”
19 November 2021, the FDA again reissued their EUA for the Pfizer-BioNTech vaccine
· “There is no adequate, approved, and available alternative Pfizer-BioNTech COVID‑19 Vaccine to prevent COVID-19.”
· “Although COMIRNATY® (COVID-19 Vaccine, mRNA) is approved to prevent COVID-19 in individuals 16 years of age and older, there is not sufficient approved vaccine available for distribution to this population in its entirety at the time of reissuance of this EUA.”
· “The Fact Sheet for Recipients and Caregivers was updated as the Vaccine Information Fact Sheet for Recipients and Caregivers, which comprises the Fact Sheet for the authorized Pfizer-BioNTech COVID-19 Vaccine and information about the FDA-licensedvaccine, COMIRNATY (COVID-19 Vaccine, mRNA).”
· The revised Pfizer-BioNTech Fact Sheet states: “The recipient or their caregiver has the option to accept or refuse Pfizer-BioNTech COVID-19 Vaccine.” This language in the fact sheet is a condition of the FDA’s granting an EUA, required for administering an EUA vaccine being distributed to potential recipients (including Service Members).
· “Drug establishments are required to provide FDA with a current list of all drugs manufactured, prepared, propagated, compounded or processed for sale in the U.S. at their facilities. Drugs are identified and reported using a unique, three-segment number called the National Drug Code (NDC) which serves as the FDA’s identifier for drugs. FDA publishes the listed NDC numbers in the NDC Directory which is updated daily.”
· “The Purple Book is a database that contains information about all FDA-licensed biological products regulated by the Center for Drug Evaluation and Research (CDER), including licensed biosimilar and interchangeable products, and their reference products.”
· The only vaccine currently in the FDA Purple Book for treatment of COVID-19 is the BLA licensed #125742 Comirnaty®. NDC# 0069-2025-01
· Every FDA letter of authorization, Pfizer-BioNTech, Moderna, & Janssen all include the statement that: “There is no adequate, approved, and available alternative” and that ‘Although Comirnaty (COVID-19 Vaccine, mRNA) is approved to prevent COVID-19 in certain individuals …., there is not sufficient available, approved vaccine, as the approved vaccine is not available for distribution to this population in its entirety at the time of reissuance of this EUA’
· Pfizer-BioNTech: “Although COMIRNATY® (COVID-19 Vaccine, mRNA) is approved to prevent COVID-19 in individuals 16 years of age and older, there is not sufficient approved vaccine available for distribution to this population in its entirety at the time of reissuance of this EUA.”
· Moderna: “Although Comirnaty (COVID-19 Vaccine, mRNA) is approved to prevent COVID-19 in certain individuals within the scope of the Moderna COVID-19 vaccine authorization, there is not sufficient available, approved vaccine, as the approved vaccine is not available for distribution to this population in its entirety at the time of reissuance of this EUA
· Janssen: “Although Comirnaty (COVID-19 Vaccine, mRNA) is approved to prevent COVID-19 in individuals within the scope of the Janssen COVID-19 vaccine authorization, there is not sufficient available, approved vaccine, as the approved vaccine is not available for distribution to this population in its entirety at the time of reissuance of this EUA”
What does the CDC say about the approved Comirnaty?
o “COMINARTY products are not orderable at this time. NDCs are listed per FDA Structured Product Label (SPL) document for the BLA licensed product. These codes are not included in CDC Vaccine Code Set files at this time. Pfizer has provided the following statement regarding the COMINARTY branded NDCs and labels: “Pfizer received FDA BLA license on 8/23/2021 for its COVID-19 vaccine for use in individuals 16 and older (COMIRNATY). At that time, the FDA published a BLA package insert that included the approved new COVID-19 vaccine tradename COMIRNATY and listed 2 new NDCs (0069-1000-03, 0069-1000-02) and images of labels with the new tradename.At present,Pfizer does not plan to produce any product with these new NDCs and labels over the next few months while EUA authorized product is still available and being made available for U.S. distribution. As such, the CDC, AMA, and drug compendia may not publish these new codes until Pfizer has determined when the product will be produced with the BLA labels.”
What does the NIH say about the approved Comirnaty?
o “Pfizer received FDA BLA license on 8/23/2021 for its COVID-19 vaccine Posted: September 13, 2021
§ Pfizer received FDA BLA license on 8/23/2021 for its COVID-19 vaccine for use in individuals 16 and older (COMIRNATY). At that time, the FDA published a BLA package insert that included the approved new COVID-19 vaccine tradename COMIRNATY and listed 2 new NDCs (0069-1000-03, 0069-1000-02) and images of labels with the new tradename.
§ At present, Pfizer does not plan to produce any product with these new NDCs and labels over the next few months while EUA authorized product is still available and being made available for U.S. distribution. As such, the CDC, AMA, and drug compendia may not publish these new codes until Pfizer has determined when the product will be produced with the BLA labels.”
· The NIH has an archived record of the 23 August 2021 approved labeling for Comirnaty NDC 0069-1000-03, & 02 for packaged vials, and 0069-1000-01 for individual vials.
I have concluded that the emergency use of Moderna COVID‑19 Vaccine for the prevention of COVID-19 when administered as described in the Scope of Authorization (Section II) meets the criteria for issuance of an authorization under Section 564(c) of the Act, because:
A. SARS-CoV-2 can cause a serious or life-threatening disease or condition, including severe respiratory illness, to humans infected by this virus;
B. Based on the totality of scientific evidence available to FDA, it is reasonable to believe that Moderna COVID‑19 Vaccine may be effective in preventing COVID-19, and that, when used under the conditions described in this authorization, the known and potential benefits of Moderna COVID‑19 Vaccine when used to prevent COVID-19 outweigh its known and potential risks; and
C. There is no adequate, approved, and available alternative to the emergency use of Moderna COVID-19 Vaccine to prevent COVID-19.
· “Although SPIKEVAX (COVID-19 Vaccine, mRNA) and Comirnaty (COVID-19 Vaccine, mRNA) are approved to prevent COVID-19 in certain individuals within the scope of the Moderna COVID-19 Vaccine authorization, there is not sufficient approved vaccine available for distribution to this
population in its entirety at the time of reissuance of this EUA.”
· To date (16 Feb 2022) Spikevax is not listed on the NIH website, nor is there a publication containing NDC numbers, or labeling information.
· Example of non-interchangeability of the Spikevax vaccine:
o “Each package of the biological product is plainly marked with— (i) the proper name of the biological product contained in the package; (ii) the name, address, and applicable license number of the manufacturer of the biological product; and (iii) the expiration date of the biological product.”
(3) The term ‘‘interchangeable’’ or ‘‘interchangeability’’, in reference to a biological product that is shown to meet the standards described in subsection (k)(4), means that the biological product may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
(4) The term ‘‘reference product’’ means the single biological product licensed under subsection (a) against which a biological product is evaluated in an application submitted under subsection (k).
· (k) Licensure of biological products as biosimilar or interchangeable
(6) Exclusivity for first interchangeable biological product Upon review of an application submitted under this subsection relying on the same reference product for which a prior biological product has received a determination of interchangeability for any condition of use, the Secretary shall not make a determination under paragraph (4) that the second or subsequent biological product is interchangeable for any condition of use until the earlier of— (A) 1 year after the first commercial marketing of the first interchangeable biosimilar biological product to be approved as interchangeable for that reference product
· “Under the BPCI Act, a sponsor may seek approval of a “biosimilar” product under new section 351(k) of the PHS Act. A biological product may be demonstrated to be “biosimilar” if data show that the product is “highly similar” to the reference product notwithstanding minor differences in clinically inactive components and there are no clinically meaningful differences between the biological product and the reference product in terms of safety, purity and potency.”
· “In order to meet the higher standard of interchangeability, a sponsor must demonstrate that the biosimilar product can be expected to produce the same clinical result as the reference product in any given patient and, for a biological product that is administered more than once, that the risk of alternating or switching between use of the biosimilar product and the reference product is not greater than the risk of maintaining the patient on the reference product. Interchangeable products may be substituted for the reference product by a pharmacist without the intervention of the prescribing health care provider.”
· To date (16 Feb 2022) there have been exactly TWO drugs that have been approved for Interchangeable status 351(k): Cyltezo (adalimumab-adbm), & its reference product Humira (adalimumab) for Cyltezo’s approved uses. This occurred on 18 Oct, 2021.
· Biosimilar and Interchangeable products are all BLA approved products, along with their BLA approved reference. Again, BLA approved products are “Biological Products” as defined by the PHS Act, Sec 351.
o Sec. 564 (c)(3) ‘‘that there is no adequate, approved, and available alternative to the product for diagnosing, preventing, or treating such disease or condition”
o Sec. 564(e)(1)(A)(ii)(III) “that individuals to whom the product is administered are informed—of the option to accept or refuse administration of the product”
o Sec. 564(e)(2)(B)(i) “If the authorization under this section regarding the emergency use authorizes a change in the labeling of the product, but the manufacturer of the product chooses not to make such change, such authorization may not authorize distributors of the product or any other person to alter or obscure the labeling provided by the manufacturer.”
o "WAIVER BY THE PRESIDENT— In the case of the administration of a product authorized for emergency use under section 564 of the Federal Food, Drug, and Cosmetic Act to members of the armed forces, the condition described in section 564(e)(1)(A)(ii)(III) of such Act and required under paragraph (1)(A) or (2)(A) of such section 564(e), designed to ensure that individuals are informed of an option to accept or refuse administration of a product, may be waived only by the President only if the President determines, in writing, that complying with such requirement is not feasible, is contrary to the best interests of the members affected, or is not in the interests of national security.”
§ Provides liability for everyone in the chain of development, production, distribution, and administration of the covered countermeasures (COVID-19 vaccines)
· knowingly without legal or factual justification; and
· in disregard of a known or obvious risk that is so great as to make it highly probable that the harm will outweigh the benefit. All three of these conditions must be proven with clear and convincing evidence.
o This means you have no recourse for any damages caused by voluntarily taking the EUA vaccines
DoD Service Members have received orders with the following language
§ You are hereby ordered to become fully vaccinated with a COVID-19 vaccine that has received full licensure from the Food and Drug Administration (FDA), in accordance with FDA-approved labeling and guidance NLT DD MMM YYYY. This is a lawful order. Failure to obey this order may result in punitive or adverse administrative action.
§ Voluntary immunization with a COVID-19 vaccine under FDA Emergency Use Authorization or World Health Organization Emergency Use Listing in accordance with applicable dose requirements prior to, or after receiving this order, constitutes compliance with this order.
What do the Regulations say about mandating vaccinations?
§ Under 21 USC 564 (The Food, Drug, and Cosmetic Act), some drugs, vaccines, or devices that have not been approved or licensed by the FDA through the regular drug approval process (or not approved for an intended use) may be used as medical countermeasures to chemical, biological, radiological, and nuclear (CBRN) agents or threats, if the FDA grants an EUA
§ The Secretary of DHHS declares an emergency based on the Secretary of Defense’s determination.
o There is no adequate, approved, and available alternative medical countermeasure.
§ “The FDA may decide that potential recipients of a drug under an EUA should have the option to refuse it. The President may waive this option for military personnel.”
AR 40-562, Chapter 8, Vaccines and Other Products Used Under Emergency Use Authorization:
§ (i) Section 8-1 General: “Under 21 USC 564 (The Food, Drug, and Cosmetic Act), some drugs, vaccines, or devices that have not been approved or licensed by the FDA through the regular drug approval process (or not approved for an intended use) may be used as medical countermeasures to chemical, biological, radiological, and nuclear (CBRN) agents or threats, if the FDA grants an EUA. This EUA authority is an alternative to the otherwise applicable requirement to file an IND application and follow IND rules (see chap. 7) to use such unapproved drugs as CBRN medical countermeasures.”
§ (ii) Section 8-2 Criteria: “In general, the FDA may grant an EUA for up to 12 months, with potential renewal, based on the following: (2) There is no adequate, approved, and available alternative medical countermeasure.”
§ (iii) Section 8-3 Refusal Options: “The FDA may decide that potential recipients of a drug under an EUA should have the option to refuse it. The President may waive this option for military personnel.”
§ (iv) Section 8-6 Information requirements for emergency use authorization products: “Any recipient of an EUA vaccine or chemoprophylaxis product must receive the information (for example, briefing, individual counseling, information statements) required by the FDA-approved EUA. Full compliance with this requirement is critical.”
§ The Proponent and exception authority is The Surgeon General, and “The proponent has the authority to approve exceptions or waivers to this regulation that are consistent with controlling law and regulations. The proponent may delegate this approval authority, in writing, to a division chief within the proponent agency or its direct reporting unit or field operating agency, in the grade of colonel or the civilian equivalent. Activities may request a waiver to this regulation by providing justification that includes a full analysis of the expected benefits and must include formal review by the activity’s senior legal officer. All waiver requests will be endorsed by the commander or senior leader of the requesting activity and forwarded through their higher headquarters to the policy proponent”
Table C–1 (Medical exemption codes)
o ‘MS’, Medical, supply, Exempt due to lack of vaccine supply. Up to 90 days
o ‘MD’, Medical, declined, Declination of optional vaccines
o 5.2.1. May, if at the time of the need under a force health protection program for a medical countermeasure against a particular threat, no satisfactory FDA-approved medical product is available, request approval by the ASD(HA) to use an unapproved product under an EUA or, if an EUA is not feasible, under an IND application. Such requests must:
o 5.2.1.1. Be justified based on the available evidence of the safety and efficacy of the medical product and the nature and degree of the threat to personnel.
o 5.2.1.2. Document a high threat for which the use of a drug under an EUA or IND application is needed, consideration of the risks and benefits of use of the drug involved, and compliance with the requirements of this Instruction.
o 5.2.1.3. Be coordinated with the Chairman of the Joint Chiefs of Staff (and if from the Commander of a Combatant Command, be submitted through the Chairman of the Joint Chiefs of Staff), the Secretary of the Army as Lead Component, and the General Counsel of the Department of Defense.
o 5.2.2. Shall, when requesting approval to use a medical product under an EUA or IND application, develop, in coordination with the Secretary of the Army, medical protocols, compliant with this Instruction, for use of the product and, if the request is approved, execute such protocols in strict compliance with their requirements.
o 5.2.3. Shall, when using medical products under a force health protection program pursuant to an EUA, comply with Enclosure 3, Federal Food Drug and Cosmetic Act section 564 (Reference (d)), section 1107a of Reference (e) and applicable FDA requirements.
o 5.2.4. Shall, when using medical products under a force health protection program pursuant to an IND application, comply with Enclosure 4, section 1107 10 U.S.C., and applicable provisions of References (e) through (g). Requirements applicable to the use of medical products under an IND application do not apply to the use of medical products under an EUA within the scope of the EUA.
o 5.2.5. May, unless otherwise provided by ASD(HA), make available to Emergency Essential civilian employees, consistent with DoD Directive 1404.10 (Reference (h)), and/or contractor personnel accompanying the Armed Forces, consistent with DoD Instruction 3020.41 (Reference (i)), who are subject to the same health risk the medical products provided under an EUA or IND application to military personnel under the same terms and conditions, except that the authority to waive an option to refuse under section 1107a of Reference (e) or informed consent under section 1107 of Reference (e) is inapplicable to these personnel.
· REQUIREMENTS AND PROCEDURES APPLICABLE TO EUAs:
o E3.2. Request for EUA. Upon or in anticipation of a declaration of emergency referred to in section E3.1, the ASD(HA) may request from the Commissioner of Food and Drugs an EUA for use of a medical countermeasure within the scope of the declaration of emergency. The request for EUA shall comply with requirements of section 564 of Reference (d) and other requirements of the FDA. Combatant Commanders, through the Chairman of the Joint Chiefs of Staff, and other heads of DoD Components may recommend to the ASD(HA) the submission of requests under this paragraph.
o E3.3. Implementation of EUA. DoD Components using medical products under an EUA shall comply with all requirements of section 564 of Reference (d), FDA requirements that are established as a condition of granting the EUA (except as provided in section E3.4 concerning a waiver of an option to refuse), guidance from the Secretary of the Army as Lead Component, and instructions from the ASD(HA).
o E3.4. Request to the President to Waive an Option to Refuse. In the event that an EUA granted by the Commissioner of Food and Drugs includes a condition that potential recipients are provided an option to refuse administration of the product, the President may, pursuant to section 1107a of Reference (e), waive the option to refuse for administration of the medical product to members of the armed forces. Such a waiver is allowed if the President determines, in writing, that providing to members of the armed forces an option to refuse is not in the interests of national security. Only the Secretary of Defense may ask the President to grant a waiver of an option to refuse.
o E3.4.1. Combatant Commanders, through the Chairman of the Joint Chiefs of Staff, and other heads of DoD Components may recommend to the Secretary of Defense, through the ASD(HA), that the Secretary request a Presidential waiver of an option to refuse administration of an EUA product.
o E3.4.2. If the President waives an option to refuse, DoD Components shall comply with all other EUA requirements, including the requirement for information provided to recipients of the EUA product consistent with section 1107a(b) of Reference (e).
Vaccine Labels:
· The approved label includes the appropriate license number No. 2229. See below for image, including the appropriate License and NDC numbers, and exclusion of the “For use under Emergency Use Authorization”
· The EUA versions of the vaccine do not have the required Trade Name COMIRNATY, US License No. 2229 or manufacturing facility addresses. They also have their own unique NDC #s:
· The labels for Moderna and Janssen “For use under Emergency Use Authorization”
There is currently no labeling published for the Spikevax vaccine
Questions in regard to this document or the facts contained within can be directed to:
























https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=23e93ef9-c51c-4e13-9cb3-e65b6a0f1968
The alibi spikevax decoy labelling is up by the way. Thanks for the great write-up!
This is an excellent post thank you